HC Certification process in Canada
I. Introduction to HC
Health Canada requires all manufacturers of medical devices sold into the Canadian market to be registered with the Canadian Medical Devices Conformity Assessment System (CMDCAS) to demonstrate compliance with Canadian medical device regulations.
Canada follows a system that combines government registration with third-party quality system review - a third-party organisation accredited by the Standards Council of Canada (SCC) to conduct CMDCAS audit.
All medical devices, whether produced locally in Canada or imported, that are intended for sale in the Canadian market, must be licensing by Health Canada, Canada's medical device authority, for evaluation under CMDCAS.
II. Applicable Regulations
Medical Devices Regulations SOR/98-282
III. Medical device classification
According to the Canadian Medical Devices Regulations (CMDR) SOR/98-282 as published by Health Canada, medical devices are classified into four categories (I, II, III, and IV) based on their level of risk associated with their use. Class I devices have the lowest risk, while Class IV devices pose the highest risk.
IV. HC Certification information
Class I medical devices are exempt from registration. The registration requirements for Class II, III, and IV devices are as follows:
1. General Registration Information:
(1) Name of the device.
(2) Classification of the device.
(3) Identification of the device.
(4) Name and address of the manufacturer as it appears on the product label.
(5) If the manufacturing location is different with (4), then the application should include the name and address of that manufacturing location.
2. Additional information required for registration of Class II medical devices:
(1) A description of the device's intended purpose and use in medical conditions for manufacturing, sale, or representation.
(2) A list of standards that the device complies with to meet safety and effectiveness requirements.
(3) A declaration of conformity with safety and effectiveness made by the senior management of the manufacturer.
(4) A declaration of conformity with Canadian medical device regulations for device labeling made by the senior management of the manufacturer.
(5) If it is an in vitro diagnostic device for near-patient testing (i.e., intended for use outside of a hospital and in a home setting), a declaration from the senior management of the manufacturer that testing has been conducted with a representative substance from the human body under conditions similar to the expected usage.
(6) A system certificate (CAN/CSA-ISO 13485:2003) issued by a CMDCAS-recognized organization.
3. Additional requirements for registration of Class III medical devices:
(1) A description of the device and the materials used in its manufacturing and packaging.
(2) A list of countries where the device has been approved for sale, along with quantities sold and any reported issues or recalls.
(3) If the device is sold as sterile, a description of the sterilization method.
(4) A description of research conducted by the manufacturer for safety and effectiveness, along with the conclusions drawn from the research.
(5) Copies of the device label.
(6) If it is an in vitro diagnostic device for near-patient testing, information about testing with a representative substance from the human body under conditions similar to the expected use.
(7) Literature references to all publicly available reports related to use, safety, and effectiveness.
4. Additional requirements for registration of Class IV medical devices:
(1) Risk assessment, including risk analysis, risk evaluation, and measures to mitigate risks to ensure safety and effectiveness.
(2) Quality plan related to the device, including specific quality practices, resources, and action procedures.
(3) Parameters of materials used in manufacturing and packaging.
(4) A list of standards adopted during design and manufacturing to meet safety and effectiveness requirements.
(5) Detailed information on all research conducted by the manufacturer to demonstrate compliance with safety and effectiveness requirements, including:
i) Pre-clinical studies and clinical studies;
ii) Process validation studies;
iii) When applicable, software validation studies;
iv) Literature studies.
(6) Objective evidence of biological safety for devices that are not in vitro diagnostic devices and are derived from or come into contact with animal tissues.
(7) Detailed information about research testing with representative substances from the human body under similar usage conditions for in vitro diagnostic devices intended for near-patient testing.
(8) Conclusions drawn by the manufacturer based on the research in (5).
A summary of the research conducted in (6) and the conclusions drawn from it.
V. System assessment
Class I medical devices in Canada are exempt from full registration, but they still require a Medical Device Establishment License (MDEL) to be sold within Canada. Health Canada conducts routine and active assessments of companies holding MDEL, which can include three types of activities:
1. Manufacturing Class I medical devices.
2. Importing Class I to IV medical devices.
3. Distributing Class I to IV medical devices.
For Class II, III, and IV medical devices, a product review and registration process is necessary. The registration timeline is 15 business days, 75 business days for Class III products, and 90 business days for Class IV products. This process is referred to as a Medical Device License (MDL).
Manufacturers of Class II, III, and IV medical devices must submit a Medical Device Quality Management System Certification from a third-party organization accredited by the CMDCAS. This certificate, CAN/CSA–ISO 13485, is one of the required documents for the registration application. Therefore, companies planning to register products in Canada must undergo a medical device quality management system audit before applying for registration.
Previously, for Class II to IV medical devices, Canada recognized quality system certificates issued by registrars under the CMDCAS. However, since January 1, 2019, CMDCAS has been completely replaced by the Medical Device Single Audit Program (MDSAP).
Authorized audit organizations, including Health Canada and other regulatory bodies, assess manufacturers of Class II to IV medical devices through separate procedures, known as the MDSAP, with systems required to comply with ISO 13485:2016.
Assessments of foreign companies can be conducted in-person or remotely from Canada. When remote, facilities and interviews with personnel can be checked using recordings, video conferences, and telephone interviews.
After the medical device license is issued, manufacturers must submit a reconfirmation to Health Canada annually on November 1st. Requests for canceling a manufacturing license should be made within 30 days of ceasing sales in Canada.
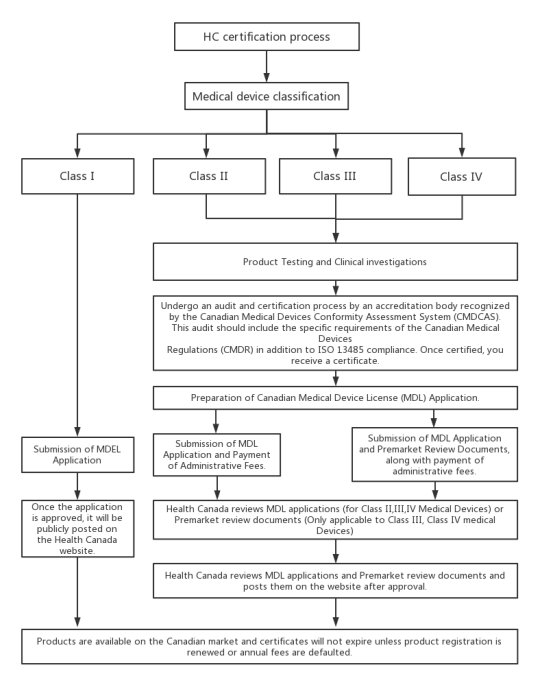
VI. HC Certification process.
1. Class I:
(1) Preparation of Technical Documentation for obtaining a Medical Device Establishment License (MDEL):
① Review official guidelines.
② Determine if the product qualifies as a medical device.
③ Confirm if the product meets the criteria for the MDEL pathway.
④ Apply for small business fee reduction (applicable both domestically and internationally).
⑤ Complete the application form.
(2) Submission of MDEL Application and Payment of Administrative Fees.
(3) Once the application is approved, it will be publicly posted on the Health Canada website.
2. Class II:
(1) Undergo an audit and certification process by an accreditation body recognized by the CMDCAS. This audit should include the specific requirements of the Canadian Medical Devices Regulations (CMDR) in addition to ISO 13485 compliance. Once certified, you receive a certificate.
(2) Preparation of Canadian Medical Device License (MDL) Application.
(3) Submission of MDL Application and Payment of Administrative Fees.
(4) Health Canada reviews the MDL application. If approved, the information is published on the Health Canada website.
3. Class III and IV:
(1) Undergo an audit and certification process by an accreditation body recognized by the CMDCAS. This audit should include the specific requirements of the Canadian Medical Devices Regulations (CMDR) in addition to ISO 13485 compliance. Once certified, you receive a certificate.
(2) Preparation of Canadian Medical Device License (MDL) Application.
(3) Submission of MDL Application and Premarket Review Documents, along with payment of administrative fees.
(4) Health Canada reviews the MDL application and the premarket review documents. If approved, the information is published on the Health Canada website.


「Med Goabroad」Official Account