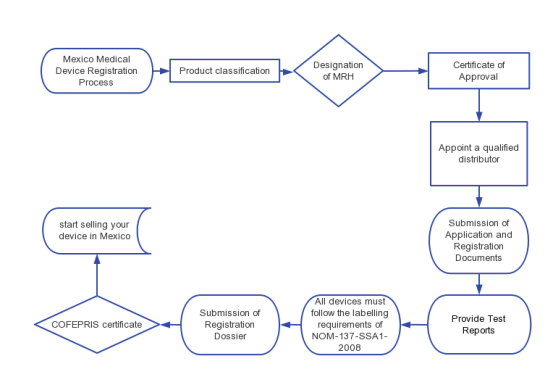
Mexico Medical Device Registration Process
I. Introduction to COFEPRIS
The Mexican regulatory body that regulates medical devices is the Federal Commission for the Defence of Health Risks (Comisión Federal para la Protección contra Riesgos Sanitarios, COFEPRIS), which is under the jurisdiction of the Mexican Ministry of Health. It regulates medical devices and in vitro diagnostic devices (IVD). In addition to medical devices, COFEPRIS also oversees drug regulation, food safety and environmental protection.
II. Regulations
1. Basic framework: Mexico's General Health Law (Ley General de Salud)
2. Good Manufacturing Practice for Medical Devices: NOM-241-SSA1-2012.
3. Medical device product labeling regulations: NOM-137-SSA1-2008.
4. Technical Vigilance Programme: lNOM-240.
III. Classification of medical devices
According to NOM-241-SSA1-2012 and the risk level of device, Mexican medical devices are classified as low risk, Class I, Class II, Class III, of which low risk devices do not require registration.
For Class I, Class II, Class III devices need product registration, but there is no system requirement and GMP will not be audited.
Class I: Conventional medical devices, the use of which safety is guaranteed and which are not implanted in the human body.
Class II: Implantable medical devices widely used in medical practice (production materials may vary), and their retention time in the human body is less than 30 days.
Class III: New or recently put into use medical devices, or the retention time in the body more than 30 days medical devices.
IV. Medical device registration information
1. Free sale certificate FSC or CFG
2. ISO13485 quality system certificate
3. Authorisation letter (fixed format, double certification is required)
4. Clinical evaluation report or clinical trial report
5. Instructions for use and labeling
6. Safety test report
7. Other technical documents and some system documents.
The above documents need to provide a Spanish version
The registration period will vary depending on the category, probably between 4-10 months. The registration certificate is valid for 5 years.
V. System Assessment
1. Class I: There is no need to go through a detailed equipment review process, a simple notification is sufficient to sell the device in Mexico. Documentation Requirements:
Authorisation letter
Labeling of the product
Instruction for use
Representative agreement
Distribution letter from each distributor
Free sale certificate (the manufacturer can sell the product in China and export it abroad)
Authorisation from the Ministry of Health of China.
2. Three routes for certification of Class I, II and III:
(1) Standard: A full set of technical documents that must be submitted and receive a full-cycle audit compared to the equivalent route.
(2) Equivalent Protocol Route: This applies to all medical devices approved in the United States. Or, if it has been approved in Canada or Japan, but is not Class I in those countries. If it is Class I medical device, then you have to choose the Standard or Standard + Third Party route. Fewer technical documents must be submitted than with the standard process, and the review time will be much shorter.
(3) Standard + Third Party Route: This only applies to new registrations or any other modifications and is newer (does not apply to the equivalent route). This option cannot be used if the process has been previously started through the equivalence agreement route. COFEPRIS endorses the use of Third Party Review (TPR). TPR is often a commercial private organisation authorised by COFEPRIS to initially review the application. If all sections meet their criteria, they will write a formal technical report for COFEPRIS recommending approval. Often, after the TPR submits their report, no additional information is required to be submitted to COFEPRIS, thus speeding up the approval time. However, this service tends to have a higher cost.
VI. Mexico medical device approval process
1. Product classification
Determine whether your medical device or IVD is on the COFEPRIS deregulated product list. If it is not in the list, please determine the classification of the device according to the rules in the Medical Device Classification Standard (Appendix II of the Medical Device Supplement of the Mexican Pharmacopoeia).
2. Designation of MRH
For all categories: If you do not have a company in Mexico, appoint a Mexican Registration Holder (MRH) to act as your local representative. Your MRH manages your medical device application and controls your medical device/IVD registration in Mexico. The MRH requirement is that it must be a local business, but does not require a unique binding. A manufacturer can have multiple licensees registered at the same time. The cost of registration is approximately 10,000 pesos, which is roughly 1,000-2,000 RMB, which is relatively low.
3. Approval Certificate
For all categories: Proof that your device is approved by the country of origin. * A common way to fulfill this requirement is to use a Certificate of Free Sale (CFS) or a Certificate to Foreign Government (CFG).
4. Distributors
For all categories: Appoint a qualified distributor to bring your medical device or IVD into Mexico. Prior to submission, you should list your distributor in your registration file to avoid the need to amend your registration at a later date.
5. Submission of Application and Registration Documents
For Class I Low Risk Medical Devices: Submit an application to COFEPRIS that includes basic information about the company and the medical device. All documents must be submitted in Spanish.
For Class I, II and III devices: Prepare a detailed registration dossier including device/manufacturing information, lab test reports, etc. Provide evidence of compliance with quality management requirements (e.g. ISO 13485 certificate) and/or CE certificate. --or-.
If your device is registered in the United States, Canada or Japan, your device may be eligible for an equivalent review pathway, which has relatively few documentation requirements. *
6. Provide Test Reports
For Class I, II and III devices: You may be required to provide specific test reports, depending on the function and intended use of the product. Generally, reports from tests conducted outside of Mexico in accordance with international standards are acceptable.
7. Labeling Requirements
For all categories: All devices must follow the labeling requirements of NOM-137-SSA1-2008. Labels and instructions must be in Spanish.
8. Submission of Registration Dossier
For Class I, II and III devices: MRH submits the registration dossier to COFEPRIS or a third party reviewer** for review and pays the registration fee. All files must be submitted in Spanish.
9. Registration Approval
For all categories: COFEPRIS issues a certificate and publishes proof of registration on the COFEPRIS website. The Certificate of Registration is valid for 5 years. Some products may require an import licence to enter Mexico.
10. Selling
Now you can start selling your device in Mexico. Update your reporting procedures to comply with the Mexican Technical Vigilance Requirements under NOM-240-SSA1-2012.


「Med Goabroad」Official Account