Indonesia Medical Device Registration Process
I. Introduction of MOH
Medical devices in Indonesia are regulated by the National Agency for Drug and Food Control (NADFC), a department within the Indonesian Ministry of Health (MOH). The MOH regulates the sale of medical devices based on the Regulation of the Ministry of Health of the Republic of Indonesia No. 1191/MENKES/PER/VIII/2010. According to Article 9, the company selling medical devices in Indonesia should be a local entity company and have a Medical Device Dealer License (IzinPenyalur Alat Kesehatan, IPAK) issued by the Ministry of Health of Indonesia.
II. Regulations
1. 20201221-Regulation on Supervision and Administration of Medical Devices
2. 20141001-Measures for the Administration of Medical Device Registration
3. 20141001-Medical Device Instructions and Labeling Regulations
4. 20140530-CFDA on the release of medical device product technical requirements for the preparation of guiding principles of the circular
5. 520141001-CFDA on the publication of medical device registration declaration information requirements and approval of the format of the supporting documents notice
III. Classification
Medical devices are categorized into four risk classes in Indonesia, and manufacturers are required to classify each medical device according to its intended use. Class A has the lowest risk class, while Class D has the highest risk class.
Classification | Risk Level |
A | Low |
B | Low-Medium |
C | Medium-High |
D | High |
IV. MOH Certification Information
The application for registration must be submitted in accordance with the ASEAN CSDT.
ASEAN CSDT includes:
1. Executive Summary
2. Equipment labeling
3. Detailed manufacturer information
4. Basic principles of medical device safety and performance and methods used to demonstrate compliance
5. Summary of design verification and validation documents
6. Risk analysis
7. Destruction methods
8. Quality management certification
9. Letter of certification of intended use/instructions/packaging, letter of certification of labeling from the manufacturer or product owner, and instructions for use
10. Declaration of Conformity
11. Manufacturer's market history certification confirmation letter
12. The manufacturer's safety confirmation letter
13. Proof of approval from foreign medical device regulatory authorities (although Indonesia does not have a requirement for country of origin approval, foreign manufacturers need approval from a reference country, the reference countries are: Australia, Canada, European Union, Japan, United States)
14. Authorization letter
15. Grouping instruction letter (if required)
V. System Assessment
Medical devices and IVDs must obtain a registration number and product license (AKA Marketing License) issued by the Ministry of Health to the local licensed distributor. Documents required for registration of medical devices in Indonesia the registration application must be submitted in accordance with the ASEAN Common Submission Dossier Template (CSDT).
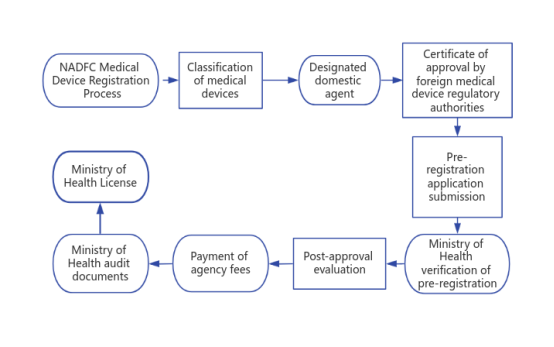
VI. NADFC Medical Device Registration Process
The Indonesian LAR must create an account in the online portal. The registration process is the same for all device classes. However, documentation requirements vary by device class. Registration is a two-stage process, namely:
1. Pre-registration process
2. Evaluation process
The Ministry of Health verifies the classification of the device and determines the cost of the assessment. The result of the pre-registration and an invoice are sent to the applicant via email. The local representative pays the fee on behalf of the manufacturer and uploads proof of payment. The Ministry of Health will review these documents and send the results to the applicant via email. Some devices require in-country testing at an accredited laboratory.
Classification | Risk Level | MOH Marketing Authorization Timeline | MOH Renewal/Change Timeline | ||
Classification process (days) | Evaluation process (days) | Classification process (days) | Evaluation process (days) | ||
A | Low | 7 | 45 | 7 | 45 |
B | Low-Medium | 7 | 90 | 7 | 45 |
C | Medium-high | 7 | 100 | 7 | 45 |
D | High | 7 | 120 | 7 | 45 |
1. Classification of medical devices
2. Designated domestic agent
3. Certificate of approval by foreign medical device regulatory authorities
4. Pre-registration application submission
5. Ministry of Health verification of pre-registration
6. Post-approval evaluation (high-risk medical devices need to submit the approval of regional regulatory agencies, device information, non-clinical data, clinical data, instructions for use and labeling, quality system documentation.)
8. Payment of agency fees
9. Ministry of Health audit documents: If the document audit passes, the applicant will get AKD/AKL certification. However, if there are problems in the document audit stage and the applicant fails to pass, the applicant will enter the revision or additional information section. If the Ministry of Health still does not accept the application after resubmission, then it must be re-registered. Previously paid PNBP cannot be refunded.
10. Ministry of Health License
11. Sale


「Med Goabroad」Official Account