
展开筛选
澳大利亚医疗器械注册流程
一、TGA介绍
根据澳大利亚医疗用品法(Therapeutic Goods Act) 规定,所有在澳大利亚上市的医疗用根据澳大利亚医疗用品法(Therapeutic Goods Act) 规定,所有在澳大利亚上市的医疗用品(药品和医疗器械)必须按有关要求,向澳大利亚医疗用品管理局(TGA)提出注册或登记申请,获得注册/登记(Australian Register of Therapeutic Goods, ARTG)后才能合法上市。
二、适用法规
1、Therapeutic Goods Act 1989-the Act;
2、Therapeutic Goods(Medical Devices)Regulations 2002-the Regulations。
三、医疗器械分类
澳大利亚是全世界仅有的将“中药”和“保健食品”直接按"药品”注册管理的发达国家,也是所有发达国家中程序清晰、标准明确和较易于获得成功注册的发达国家,通过澳大利亚药品注册/登记,举例:
类别 | 风险级别 | 器械例子 |
ClassI | 低 | 手术牵开器、压舌板 |
ClassI - 无菌 | 中低档 | 皮下注射针头,抽吸装置 |
ClassI - 包含测量功能 | 中低档 | 具有特定测量单位的药杯 |
Class Ila | 中低档 | 数字或红外线温度计 |
Class Il b | 中等偏上 | 肺呼吸机、血袋、避孕套 |
Class Ill | 心脏瓣膜、主要关节置换植入物 含有药物或组织、动物、生物或微生物来源的细胞或物质的装置 | |
AIMD(有源植入式医疗器械) | 高 | 植入式除颤器 |
四、TGA认证资料
①澳洲技术文件(Australian Technical File,ATF):医疗器械的申请人需要准备ATF,其中包括技术规格、性能数据、临床评估等信息。
②安全性和有效性数据
③质量管理体系证明
④标签和使用说明
五、体系考核
1、TGA会监控医疗器械,即使它们已获准在澳大利亚使用,也可确保它们继续满足我们的安全标准和法规要求。这就是所谓的上市后监控。一些上市后监测活动包括:
(1)评估和调查医疗设备问题报告
(2)检查医疗器械是否继续符合基本原则的证据
(3)对制造商进行定期检查
(4)要求制造商和赞助商在特定时间范围内报告不良事件和涉及其医疗设备的其他信息。
(5)审查不良事件报告只是TGA监视澳大利亚使用的治疗用品安全性的一种方式。
(6)TGA将这些报告收集到数据库中,并定期对其进行监视以识别峰值或异常趋势。TGA内的临床医生和科学家组成的小组进行了风险评估,以确定是否需要进行调查,我们也可能会获得专家的建议。这些调查可能会导致产品回收(召回),危险和安全警报,制造商对产品的修改/改进或对生产场所的监督审核。
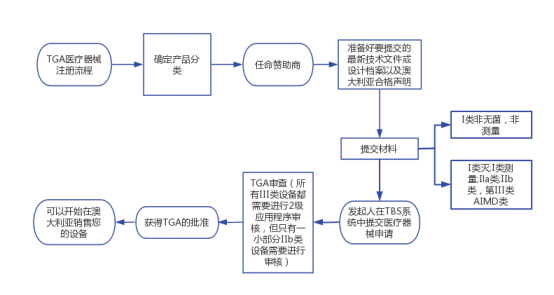
六、医疗器械TGA注册流程
1、医疗器械分类:使用2002年澳大利亚治疗用品(医疗器械)条例附表2确定分类。如果您的设备具有欧洲CE标记,则分类可能相同。作为注册的一部分,治疗用品管理局(TGA)通常会接受来自公告机构的CE标记证书。TGA还接受具有可接受的海外营销批准的MDSAP证书(例如,加拿大卫生部MDL,日本MHLW/PMDA PMC或PMA,美国FDA 510(k)或美国新FDA);或获得日本MHLW/PMDA PMC或PMA的日本MHLW/PMDA QMS认证。
2、任命赞助商:如果您在澳大利亚没有本地办事处,请任命澳大利亚TGA赞助商。赞助商可以方便您进行设备注册,充当制造商与TGA之间的联络人,并且赞助商的名称必须出现在您的设备和标签上。
3、准备资料:准备好要提交的最新技术文件或设计档案以及澳大利亚合格声明。
4、提交材料:对于除I类非无菌,非测量以外的所有设备,赞助商在TGA商业服务(TBS)系统中提交制造商的证据(例如CE标记证书),以供TGA审查和接受。
(1)I类不须灭菌和无测量功能的医疗器械注册流程:
①生产商准备技术材料和澳大利亚符合性声明;
②代理商通过TGA eBS申请加入ARTG名册;
③器械被ARTG和TGA认可并通知代理商,ARTG可能会选择上市后审查;
④代理商从eBS打印包容证书;
⑤对上市后的产品监控。
(2)其他类别医疗器械注册流程(即I类灭菌;I类测量;IIa类;IIb类,第III类,AIMD类):
①对产品进行分类,分为Ⅰ类灭菌、Ⅰ类测量功能、Ⅱa 、Ⅱb、Ⅲ类、AIMD;
②从TGA或者EU NB的合格评定的证据;
③生产商准备澳大利亚的符合性声明;
④代理商递交生产商的材料给TGA;
⑤如果申请成功,递交申请器械包含在ARTG表里;如果不成功,申请被撤回并作出修改提供更多材料;
⑥递交申请器械包含在ARTG表里,如果申请成功,器械包含在ARTG里代理商可以在澳大利亚销售(一些产品可能会被选作申请审查);如果不成功则申请被撤回并做出修订提供更多材料;
⑦产品上市后监管。
5、提交申请:发起人在TBS系统中提交医疗器械申请。该应用程序包括“预期目的”声明,分类和全球医疗设备命名法(GMDN)代码。支付申请费
6、TGA审查:作为2级应用程序审核的一部分,TGA将审查设计档案的各个部分。所有III类设备都需要进行2级应用程序审核,但只有一小部分IIb类设备需要进行审核。
7、获得批准:TGA将批准或拒绝您的申请。如果获得TGA的批准,将发布澳大利亚治疗用品注册(ARTG)清单编号(ARTG包含证书),并且您的清单将包含在TGA网站上的ARTG数据库中。
8、销售:您现在可以开始在澳大利亚销售您的设备。只要您不对将使ARTG列表无效的设备进行更改,注册就不会过期,当前的CE标记证书(如果适用)已向TGA存档,并且每年支付ARTG列表费。


「众信博达」 公众号