FDA Medical Device Registration Process
I. Introduction to FDA
FDA, full name Food and Drug Administration.
An Agency under the Department of Health and Human Services (DHHS)
1. Mission: Protect public health by ensuring the safety and efficacy of human and veterinary medicines, biological products and medical devices, as well as the safety of the food supply, cosmetics and radiation products. Responsible for regulating the manufacturing, marketing and distributing of tobacco products to protect public health and reduce tobacco use among minors.
2. Regulated products: foods (except agricultural products), drugs, medical devices, radiation products, cosmetics, vaccines, blood products, biological agents, animal feed, veterinary drugs, tobacco.
II. Regulations
1. Federal Food, Drug, and Cosmetic Act: 1976 Food, Drug, and Cosmetic Act, Medical Device Amendment
2. Safe Medical Device Act: 1995 Safe Medical Device Act
3. FDA Modernisation Act signed by President Clinton: 1997 Food and Drug Administration Modernization Act
4. Medical Device Assessment Fee and Modernisation Act: 2002 The Medical Device User Fee and Modernization Act
III. Classification of medical devices
Unlike the rule-based classification schemes used in the EU, Brazil and other markets, in the United States, medical devices are classified using an equivalence-based systematic classification method. The U.S. Food and Drug Administration (FDA) uses this methodology to classify medical devices as Class I, Class II or Class III according to the risk they pose to patients or users.
1. Class I: Low-risk devices, usually of simple design with no moving parts. The requirements applicable to Class I devices are called "general controls". Examples of Class I medical devices include adhesive bandages, scalpels, and manual stethoscopes. Most Class I medical devices can be registered directly with the FDA without the need for a license.
2. Class II: Moderate-risk devices, usually of more complex design, but whose failure is unlikely to cause immediate risk of serious injury or death. General controls are considered insufficient to regulate Class II medical devices and "special controls" are required. Examples of Class II medical devices include endoscopes, motorized wheelchairs, syringes and total joint implants. Some Class II medical devices require only registration, but most require a license.
3. Class III: High-risk devices, where failure could lead to serious medical complications, including death. Class III medical devices are subject to the premarket approval (PMA) process unless it can be demonstrated that they are "substantially equivalent" to an existing approved ("equivalent") device. Examples of Class III medical devices include pacemakers, deep brain stimulators, breast implants and heart valves.
IV. Medical device registration information
(1) Five copies of finished goods in complete packaging
(2) Structure diagram and description of the device
(3) Safety demonstration or test material on the performance and working principle of the device
(4) Introduction of manufacturing process
(5) Summary of clinical trials
(6) Instructions for use, which must describe in detail if the device has radioactive energy or releases radioactive material.
V. System assessment
Enterprises choose to FDA registration in October-December each year is the most cost-effective, register with the FDA after October 1, the registration number can continue to be used until the end of the next year, and only need to pay the annual fee for one year, the registration number can be used for an additional 3 months.
The FDA registration cycle is 1-2 weeks (after the company pays the annual fee to FDA successfully), there will be the Owner/Operator Number and Listing Number, which can be used for customs clearance directly. Equipment that has been registered but has not yet obtained a "Registration or FEI Number" can temporarily use this number as a "Registration or FEI Number" for export customs clearance. Among them, the Registration or FEI Number needs to wait for the FDA to assign.
The number is valid for one year and is renewed every October.
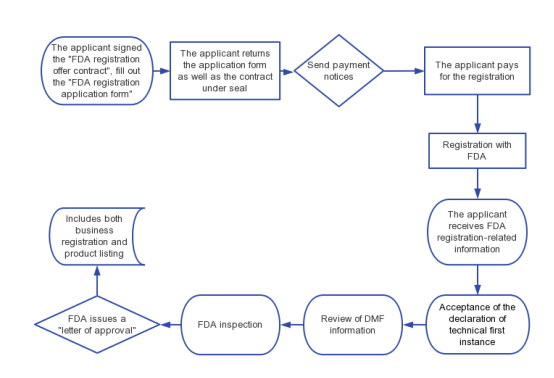
VI. FDA Registration Process of Medical Device
2. The applicant returns the application form as well as the contract under seal;
3. Send a payment notice;
4. The applicant pays for the registration;
6. The applicant receives FDA registration-related information (FDA listing Number, password, PIN code and other relevant information)
7. Acceptance of the declaration of technical first instance
8. Review of DMF information
9. FDA inspection
10. FDA issues an "approval letter".
11. Includes both business registration and product listing.
After the registration is completed, relevant information can be found by entering the listing number, search number, or operator name on the FDA.
Fees include two aspects, one is the the FDA annual fee charged by the U.S., should be paid directly to the FDA finances in the form of U.S. dollars, every year on October 1 - December 31 renewed for the next year's annual fee to maintain the effectiveness of the FDA registration, the amount of the annual fee is also different each year. (The annual fee for a Class I medical device is currently $5,546, and on average increases by several hundred dollars per year..)
The other is the agency fee charged by the consulting company (including company registration, product registration, and U.S. Authorized Representative).
Three numbers will be given after successful registration:
① Registration or FEI Number
② Owner/Operator Number
③ Listing Number
FDA Modernisation Act requires that all firms engaged in the manufacture, formulation, dissemination, synthesis, assembly, processing, or import/export of medical devices must register with the FDA.


「Med Goabroad」Official Account