印度尼西亚医疗器械注册流程
一、MOH介绍
印尼的医疗器械由印度尼西亚卫生部(MOH)里的一个部门——国家药品和食品控制局(NADFC)管理。卫生部依据印度尼西亚共和国卫生部第1191/MENKES/PER/VIII/2010号条例对医疗器械的销售进行管理。根据第9条,在印尼销售医疗器械的公司应当是当地实体公司,且拥有印尼卫生部颁发的医疗器械经销商许可证(IPAK)。
二、适用法规
1、20201221-《医疗器械监督管理条例》
2、20141001-《医疗器械注册管理办法》
3、20141001-《医疗器械说明书和标签管理规定》
4、20140530-CFDA关于发布医疗器械产品技术要求编写指导原则的通告
5、520141001-CFDA关于公布医疗器械注册申报资料要求和批准证明文件格式的公告
三、医疗器械分类
医疗器械在印度尼西亚被分为四个风险等级,制造商必须根据其预期用途将每个医疗器械进行分类。A类的风险等级最低,D类则风险等级最高。
类别 | 风险级别 |
A | 低风险 |
B | 低中度风险 |
C | 中度-高度风险 |
D | 高风险 |
四、MOH认证资料
注册申请必须按照东盟共同提交档案模板(CSDT)提交。东
盟共同提交档案模板 (CSDT) 包括:
1、执行摘要
2、设备标签
3、详细的制造商信息
4、医疗器械安全和性能的基本原则以及用于证明符合性的方法
5、设计验证和确认文件摘要
6、风险分析
7、销毁方法
8、质量管理认证
9、预期用途/指示/包装证明信、制造商或产品所有者的标签证明信和使用说明
10、符合性声明
11、制造商的市场历史证明确认信
12、制造商的安全确认信
13、外国医疗器械监管机构的批准证明(印度尼西亚虽然没有原产国批准的要求,但外国制造商需要参考国家的批准,参考国为:澳大利亚、加拿大、欧盟、日本、美国)
14、授权委托书
15、分组指示信(如果需要)
五、体系考核
医疗器械和IVD必须获得卫生部颁发给当地特许经销商的注册号和产品许可证(AKA营销许可证)。在印度尼西亚注册医疗器械所需的文件注册申请必须按照东盟共同提交档案模板(CSDT)提交。
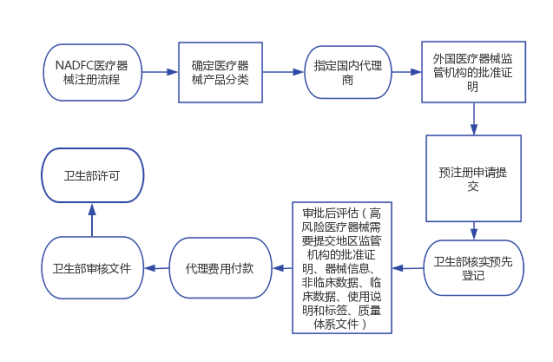
六、NADFC医疗器械注册流程
印度尼西亚LAR必须在在线门户中创建一个账户。所有设备类别的注册过程都是一样的。然而,对文件的要求因设备类别而异。注册是一个两阶段的过程,即:
1、预注册过程
2、评估过程
卫生部核实设备的分类并确定评估费用。预注册的结果和发票将通过电子邮件发给申请人。当地代表代表制造商支付费用并上传支付证明。卫生部将审查这些文件,并通过电子邮件将结果发送给申请人。有些设备需要在认可的实验室进行国内测试。
设备类别 | 风险等级 | 卫生部上市许可时间表 | 卫生部更新/变更时间表 | ||
分类流程(天数) | 评估流程(天数) | 分类流程(天数) | 评估流程(天数) | ||
A | 低风险 | 7 | 45 | 7 | 45 |
B | 低中度风险 | 7 | 90 | 7 | 45 |
C | 中度-高度风险 | 7 | 100 | 7 | 45 |
D | 高风险 | 7 | 120 | 7 | 45 |
1、医疗器械分类
2、指定国内代理商
3、外国医疗器械监管机构的批准证明
4、预注册申请提交
5、卫生部核实预先登记
6、审批后评估
7、高风险医疗器械需要提交地区监管机构的批准证明、器械信息、非临床数据、临床数据、使用说明和标签、质量体系文件
8、代理费用付款
9、卫生部审核文件: 如果文件审核阶段通过,申请人将获得AKD/AKL 认证。但是,如果文件审核阶段出现问题未通过,则将进入修改或补充资料环节。再次提交后如果卫生部仍未接受的话,那么必须重新注册。先前已付款的PNBP不能退。
10、卫生部许可
11、销售


「众信博达」 公众号